

Inference of Macroevolutionary Processes
Many evolutionary processes entail differential rates of diversification: e.g., adaptive radiation, diversity-dependent diversification, key innovations, and mass extinction. Current methods for exploring these evolutionary processes involves the use of stochastic birth-death branching process models, where rates of diversification may be (a) time-dependent, (b) lineage-specific, or (c) character-dependent. Our ultimate goal is to infer diversification rates through time and among lineages and identify correlations to genetic, phenotypic and/or environmental factors that impact diversification rate.
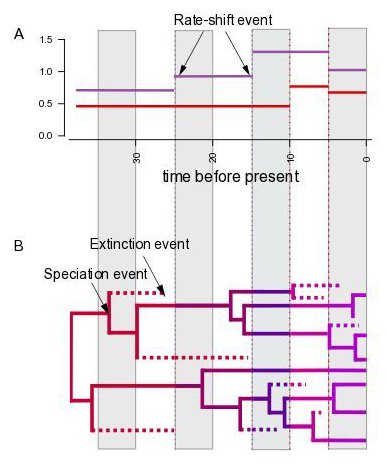
In our previous work we extended the theory on birth-death models (Höhna, 2013, Bioinformatics; Höhna, 2015, JTB; Höhna et al., 2016, Bioinformatics) and implemented computationally efficient methods to infer episodic changes in diversification rate, including cases in which species sampling is incomplete (Höhna, 2014, PLoS one; Höhna et al., 2011, MBE). In a recent study, we used this new approach to show that the diversity of conifers was impacted by one major mass-extinction event approximately 23 million years ago (May et al., 2016, MEE). This event coincides with the known timing of the increase of more arid, grassland ecosystems.
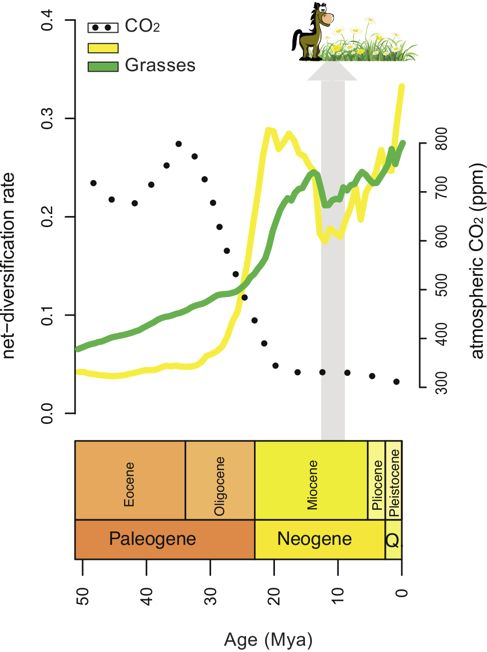
In an upcoming study, we also developed a statistical method to identify correlations between rates of diversification and environmental variables, such as changes in atmospheric CO2, which includes an empirical study of the daisy family, Asteraceae.
Most recently, we developed a new model to test whether changes chromosome number occur anagenetically or cladogenetically (Freyman and Höhna, 2018, Syst. Biol.). Interestingly, our empirical results show that chromosome numbers change anagenetically in some groups and cladogenetically in other groups. Furthermore, we developed a new stochastic character mapping algorithm to map changes in morphology coupled with speciation and extinction rates onto the phylogeny. This method enabled us to show that self-compatible provides a short-term advantage, but a long-term dead-end in Onagraceae lineages (Freyman and Höhna, 2019, Syst. Biol.).
Our next steps are to develop and implement a robust model for inferring diversification-rate variation across lineages that correct problems with previous methods (see Moore et al., 2016, PNAS). These models allow us to study diversification rate-variation through-time and among lineages. Currently, we obtain lineage-specific diversification rate estimates independently of potentially correlated factors. Future projects in this research stream include the development of statistical methods to infer correlations between time-dependent and lineage-specific diversification rates with species traits (such as morphological traits), substitution rates and environmental factors/habitat.
Finally, we want to incorporate fossil occurrence information in our models of lineage diversification. Current models that use only present-day taxa have clearly lower statistical power at inferring historical events. Molecular phylogenies combined with fossil occurrence data will provide us a unique opportunity in studying the mode of lineage diversification over time and among clades (Silvestro et al., 2018, Syst. Biol.).
References:
- WA Freyman and S Höhna: Stochastic character mapping of state-dependent diversification reveals the tempo of evolutionary decline in self-compatible Onagraceae lineages, Systematic Biology, 2018, in press
- D Silvestro, MF Tejedor, ML Serrano-Serrano, O Loiseau, V Rossier, J Rolland, A Zizka, S Höhna, A Antonelli and N Salamin: Early Arrival and Climatically-Linked Geographic Expansion of New World Monkeys from Tiny African Ancestor. Systematic Biology, 2018, in press
- FL Condamine, J Rolland, S Höhna, FAH Sperling and I Sanmartín: Testing the Role of the Red Queen and Court Jester as Drivers of the Macroevolution of Apollo Butterflies, Systematic Biology, 2018, in press
- WA Freyman and S Höhna: Cladogenetic and Anagenetic Models of Chromosome Number Evolution: A Bayesian Model Averaging Approach, Systematic Biology, 2018, 67(2), 195-215
- BR Moore, S Höhna, MR May, B Rannala and JP Huelsenbeck: Critically evaluating the theory and performance of Bayesian analysis of macroevolutionary mixtures. Proceedings of the National Academy of Sciences, 2016, 113 (34), 9569-9574
- S Höhna, MR May and BR Moore: TESS: an R package for efficiently simulating phylogenetic trees and performing Bayesian inference of lineage diversification rates, Bioinformatics, 2016, 32 (5): 789-791
- MR May, S Höhna and BR Moore: A Bayesian Approach for Detecting the Impact of Mass-Extinction Events on Molecular Phylogenies When Rates of Lineage Diversification May Vary, Methods in Ecology and Evolution, 2016, 7 (8), 947–959
- S Höhna: The time-dependent reconstructed evolutionary process with a key-role for mass-extinction events. Journal of Theoretical Biology, 2015, 380, 321-331
- S Höhna: Likelihood inference of non-constant diversification rates with incomplete taxon sampling. PLoS one, 2014, 9 (1), e84184
- S Höhna: Fast simulation of reconstructed phylogenies under global time-dependent birth-death processes. Bioinformatics, 2013, 29, 1367-1374
- S Höhna, T Stadler, F Ronquist and T Britton: Inferring speciation and extinction rates under different species sampling schemes. Molecular Biology and Evolution, 2011, 28, 2577-2589