Sonja Grath - Molecular Evolution and Functional Bioinformatics
Cold adaptation in natural populations of Drosophila
When organisms are faced with new environmental conditions, such those caused by climate change or range expansion, they must adapt in order to survive. However, it has been difficult to identify the specific genes and molecular mechanisms that underlie environmental adaption. Here we propose to investigate cold adaptation in the fruit fly Drosophila ananassae, a species that has recently expanded from its home range in tropical Asia to colonize temperate regions. We previously found that within a population of D . ananassae from Bangkok, Thailand, there is natural variation in cold tolerance, as measured by the time required for the flies to recover from a cold shock. The population otherwise does not show any sign of population structure, suggesting that the difference in cold shock recovery is determined by a small number of genes with large effects. Thus, D . ananassae is a promising model system for determining the genetic basis of cold adaptation.
 In this project, we perform high-throughput gene expression analysis of multiple D . ananassae strains before and after cold shock. We also survey genome-wide DNA sequence polymorphism in this species and use population genetic analyses to identify genes involved in cold tolerance and adaptation. Further, we conduct a preliminary QTL mapping experiment. Recombinant inbred lines will be genotyped with restriction associated DNA (RAD) tags. These data will help with the identification of genes/polymorphisms associated with adaption to cold temperatures.
In this project, we perform high-throughput gene expression analysis of multiple D . ananassae strains before and after cold shock. We also survey genome-wide DNA sequence polymorphism in this species and use population genetic analyses to identify genes involved in cold tolerance and adaptation. Further, we conduct a preliminary QTL mapping experiment. Recombinant inbred lines will be genotyped with restriction associated DNA (RAD) tags. These data will help with the identification of genes/polymorphisms associated with adaption to cold temperatures.
The results of these experiments will shed light on thermal adaptation of natural populations. In addition, the use of state-of-the-art genomic methods will provide new information on genome-wide genetic polymorphism among D. ananassae strains. These data will not only facilitate the identification of genes and polymorphisms associated with adaptation to cold, but will also uncover genes that have been subject to recent and long-term adaptive evolution.
This project is supported by a grant from the German Research Foundation (DFG, GR 4495/2-1).
DNA methylation in insects
 DNA methylation describes the process by which a methyl group is covalently attached to a DNA
DNA methylation describes the process by which a methyl group is covalently attached to a DNA
nucleotide – usually a cytosine followed by a guanine – and has been found in all domains of
life. In mammals, DNA methylation has been shown to be involved in several processes such as embryo development, genomic imprinting and gene expression regulation. However, in insects, the patterns and the functional importance of DNA methylation are less clear and seem to vary widely.
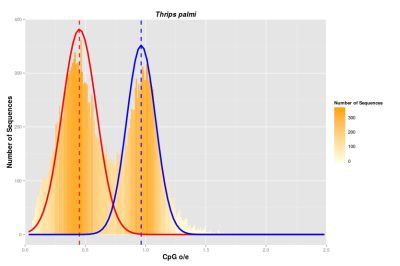
Our overall aim is to identify patterns of DNA methylation in order to test theories concerning the origin, presence and function of DNA methylation. To this end, we investigate the presence of DNA methyltransferase enzymes (DNMTs) and the distributions of CpG dinucleotides (CpG o/e) in all extant hexapod orders. To carry out the analyses, we combine both transcriptomic and genomic data.
This project is done in cooperation with scientists from the Zoological Research Museum Alexander Koenig and the Friedrich-Wilhelms-Universität in Bonn, Germany.top
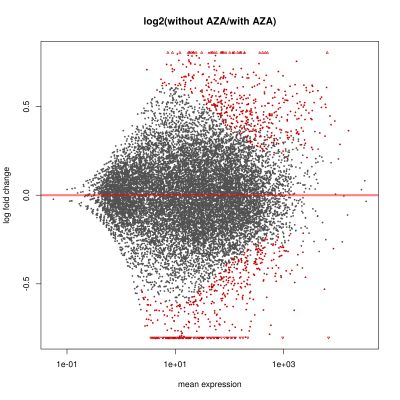
Gene expression and DNA methylation variation in acute myeloid leukemia
Cancer can be seen as evolutionary process, which is driven by mutations in somatic cells followed by subclonal selection. In the last decades, cytogenetic and candidate gene studies have identified recurrent mutations in patients with hematopoietic neoplasms. Depending on the type and complexity of the malignant transformation, multiple and genetically heterogeneous subpopulations within the cancer cell population have been observed. There is increasing evidence that these subpopulations have been generated through selection of subclones that harbour mutations conferring advantages for cell survival and growth. Intriguingly, mutations in several epigenetic regulators have been found in a broad variety of cancer types. Mutations in these regulators often lead to loss or reduction in enzymatic activity resulting in changed epigenetic landscapes and gene expression patterns. Subsequently, global DNA hypomethylation in combination with local hypermethylation is an epigenetic hallmark found in many solid and soft tissue tumours. However, it is unclear how a changed enzymatic activity of an epigenetic regulator can lead to such locus specific changes in epigenetic profiles and so far no clear correlation of these mutations and transcriptional or epigenetic changes have been found.
We currently focus on alterations in gene expression and DNA methylation patterns in two related diseases – myelodysplastic syndromes (MDS) and acute myeloid leukaemia (AML) – to better understand the general principle of epigenetic heterogeneity and its role in malignant haematopoiesis. These studies should elucidate the role of epigenetic modifiers in the evolution of hematopoietic malignancies and therapy resistance.
Projects in this research area are done in cooperation with several scientists from the CRC 1243 Cancer Evolution: Genetic and Epigenetic Evolution of Hematopoietic Neoplasms (http://www.sfb1243.biologie.uni-muenchen.de/aktuelles/new-sfb/index.html).